Auto_MD
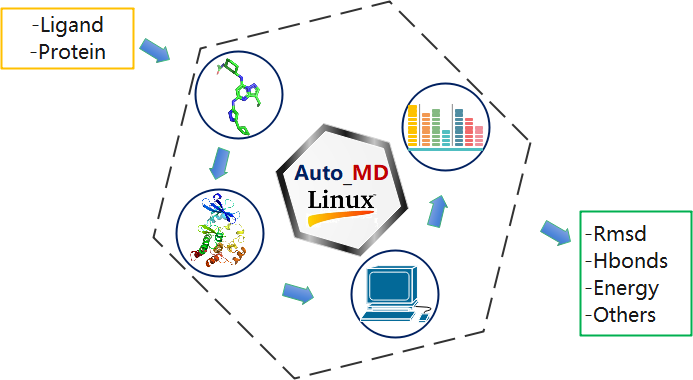
The Auto_MD module is a highly automated, high-throughput computational pipeline designed to streamline and accelerate the process of running molecular dynamics (MD) simulations using GROMACS.
This innovative module dramatically reduces the traditionally complex and time-consuming setup and submission workflow from over 30 minutes to under 1 minute. By automating every step—including system building, parameterization, solvation, energy minimization, equilibration, and production run configuration—Auto_MD eliminates manual intervention and significantly enhances the efficiency of MD ensemble construction.
Its core strength lies in enabling fully automated, batch computation of MD simulations for protein-ligand systems. Researchers can now submit multiple complexes for study with a single command, achieving unparalleled scalability.
Furthermore, Auto_MD integrates end-to-end automation, meaning a single execution not only launches the complete simulation workflow but also triggers subsequent analysis processes. This ensures that users receive finalized, ready-to-interpret results, transforming a multi-step, expert-driven task into a seamless, one-step operation for driving discovery in computational biology and drug design.